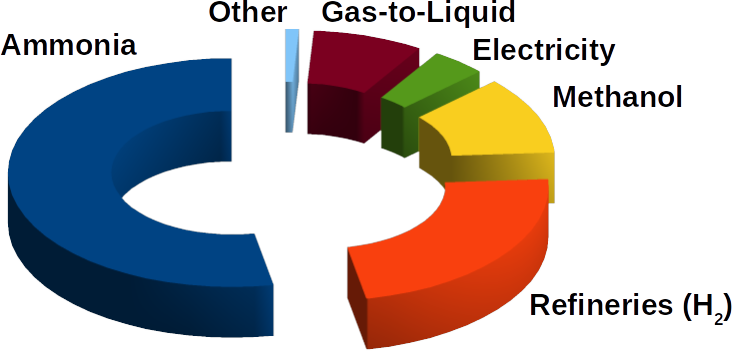
Hydrogen (H2) is a lightweight alternative fuel with a high energy density. However, its environmental impact and life cycle efficiency are determined by how it is produced. Today, the main processes of hydrogen production is either by coal gasification or steam reforming of natural gas where in the last step the produced carbon dioxide (CO2) is produced. Usually, this CO2 is released to the environment. The hydrogen produced by these processes lead is called black/brown or grey hydrogen. To improve its carbon footprint, CO2 capture is necessary. This hydrogen is then call blue hydrogen. However, to obtain zero-emission green hydrogen, electrolysis of water using renewable energy is necessary. During the electrolysis process, hydrogen and oxygen are produced on two electrodes (download our more about hydrogen production and utilization as fuel can be found in our latest DIY FC manual).
Climate-related economic pressure for more efficient gas separation processes
The produced hydrogen is not pure in any of the mentioned instances. For example, using steam methane reforming reaction there are many byproduct gases like carbon monoxide, CO2, water, nitrogen and methane gas.
Typically, the CO2 of hydrogen gas is up to 50% contributing to the greenhouse effect caused by burning fossil fuels. Currently, around 80% of CO2 emissions come from fossil fuels. It has been predicted that the concentration of CO2 in the atmosphere will increase up to 570 ppm in 2,100 which increases the global temperature of about 1.9°C.
The traditional processes of gas separation such as cryogenic distillation and pressure swing adsorption have certain disadvantages, for example high energy consumption. Therefore, developing high-quality and low-cost technologies for gas separation is an important intermediate step to produce cheap hydrogen while reducing CO2 emissions.
Application of 2D material towards gas separation
Finding low cost alternatives like membrane-based separation methods for hydrogen-CO2 separation is a potentially lucrative research and it is therefor not surprising that numerous publications have investigated the matter. The various membrane materials for gas separation range from polymeric membranes, nano-porous materials, metal–organic frameworks and zeolite membranes. The goal is to reach a good balance between selectivity and permeance of gas separation. Both are key parameters for hydrogen purification and CO2 capture processes.
A study published the journal Nature Energy by researchers of the National Institutes of Japan, offered a material platform as advanced solution for the separation of hydrogen from humid gas mixtures, such as those generated by fossil fuel sources or water electrolysis. The authors showed that the incorporation of positively charged nanodiamonds into graphene oxide (GO/ND+) results in humidity repelling and high performance membranes. The performance of the GO/ND+ laminates excels particularly in hydrogen separation compared with traditional membrane materials.
Strategy and performance of new membrane materials
Graphene oxide laminates are considered as step-change materials for hydrogen-CO2 separation as ultra permeable (triple-digit permeance) and ultra-selective membranes. Still, graphene oxide films lose their attractive separation properties and stability in humid conditions.
After lamination, graphene oxide sheets have an overall negative charge and can be disintegrated due to the electrostatic repulsion if exposed to water. The strategy to overcome this obstacle was based on the charge compensation principle. That is, the authors incorporated positively and negatively charged fillers as stabilizing agents, and tested different loadings as well as graphene oxide flake sizes. So-prepared membranes were tested for stability in dry and humid conditions while separating either hydrogen from CO2 or oxygen.
The GO/ND+ composite membranes retained up to 90% of their hydrogen selectivity against CO2 exposure to several cycles and under aggressive humidity test. A GO30ND+ membrane with 30% positively charged nano-diamond particles exhibited exceptional hydrogen permeance with more than 3,700 gas permeatin units (GPU) and high hydrogen-CO2 selectivity. Interestingly, incorporation of negatively charged nano-diamond particles had no stabilizing effect. The researcher attributed this mostly to the generation of macro scale voids in ND− systems resulting in the loss of selectivity. This phenomenon is commonly observed in polymer-based nano-composite membranes with poor interfacial interactions
The gas separation properties of the composite membranes were also investigated using an equimolar hydrogen-CO2 feed mixture. The hydrogen permeance decreased by 6% and hydrogen-CO2 selectivity of the GO30ND+ membrane by 13%.
The stability test of the membranes exposure to wet and dry feeds of the equimolar hydrogen-CO2 mixture and hydrogen-oxygen mixture showed that GO/ND+ membranes were reversible membrane properties. On the other hand, graphene oxide-only membranes could not survive a single complete cycle exposure, becoming fully permeable to both gases. The researchers explained that the advantages of GO/ND+ membranes over graphene oxide-only membranes were caused by changes of the pore architecture such as dimensions and tortuosity, which could be improved by optimizing the nano-diamond loading. This results in better permeability without any notable loss of selectivity.
X-ray diffraction analysis showed that the incorporation of nanodiamonds has two major effects on the membrane microstructure: increasing the overall pore volume and reducing the average lateral size. Both make the membrane structure more accessible for molecular transport.
Nevertheless, this relatively new class of humid-resistant membranes still needs more optimization to compete with current industrial separation processes.
Image: Pixabay / seagul