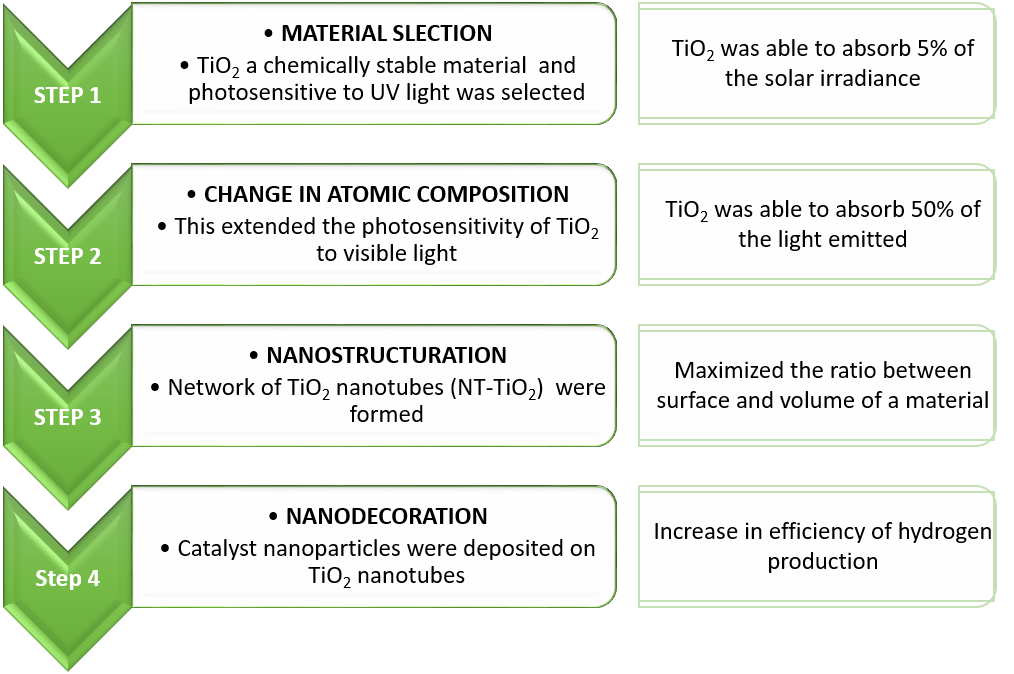
Direct ethanol fuel cells (DEFCs) are fuel cells that run on ethanol to directly produce electrical power. Despite having much to offer they have not been forayed into. Ethanol can be made from biomass by yeasts and its oxidation products – CO2 and H2O – are hence environmentally friendly. The application of DEFCs could be a lucrative solution for vehicles due to the energy efficiency if mass-produced. Our current infrastructure for combustion fuels is ready for ethanol. DEFC usage would therefore be a sustainable and environment-friendly alternative to current internal combustion engines. Moreover, ethanol is liquid, which facilitates distribution, storage and use.
According to studies sponsored by International Energy Agency (IEA), DEFCs deliver high power densities, culminating between 50 to 185 mW / cm2. Currently, DEFCs face multiple challenges such as slow redox kinetics, limited performance, and the high cost of electrocatalysts needed for DEFCs.
In a DEFC, the two key reactions are:
- Ethanol Oxidation Reaction (EOR)
- Oxygen Reduction Reaction (ORR)
Their sluggish rates have prevented widespread adoption of this technology. State-of-the-art DEFCs require expensive platinum-based materials to catalyze these reactions. Yet, they do not completely oxidize ethanol to CO2 to complete the EOR reaction, limiting the energy efficiency. One way to fix this issue is to separate and re-inject the unreacted ethanol. Since this adds more engineering to the fuel cell, a better solution is to find more efficient catalysts. Hence, to realize the true potential of DEFCs, is to find cheaper and more active catalysts for the two reactions in DEFCs.
The researchers at the University of Central Florida and their colleagues experimented on Pd–N–C catalyst and attempted to improve catalyst performance by introducing fluorine atoms. The team used alkaline membranes and platinum-free catalysts. Not only were these more cost-effective but also produced a high power output.
Previous research on electrocatalytic systems revealed that the local coordination environment (LCE) of the electrode surface is pivotal in tuning the activity of electrocatalysts made of carbon-supported metal nanoparticles. The study showed that introducing fluorine atoms in Pd–N–C catalysts regulated the LCE around the Pd, improving both activity and durability for the two key reactions. This improved the catalytic performance, and ultimately the fuel cell’s performance.
The new study demonstrated that fluorine doping rearranged the electron structure of the fuel cell catalyst. This substantially improved power density and ultimately the performance of the DEFC when compared with present-day benchmark catalysts. The experimental results on long-term stability demonstrated promising advancements towards practical applications of such catalysts in DEFCs.
Results
Upon experimental analysis, it was found that the fluorine atoms in the catalyst weakened carbon-nitrogen bond and pushed the N atoms towards palladium. This electron translocation efficiently regulated the LCE of palladium by forming palladium-nitrogen active sites for catalytic reactions.
The N-rich palladium surface promoted carbon-carbon bond cleavage and enabled complete ethanol oxidation. During the ORR, the N-rich palladium surface surface not only weakened CO2 adsorption but also created more accessible catalytic sites for rapid O2 adsorption.
According to the authors, a commonly occurring problem in DEFCs – the inability to complete the two key reactions – has been resolved. Their catalyst enhanced the overall performance of the fuel cell. The addition of fluorine also enhanced the durability of the catalyst by reducing the corrosion of carbon materials as well as inhibiting palladium migration and aggregation.
When the novel catalyst was tested in a DEFC, an output maximum power density of 0.57 W/cm2 was obtained. The fuel cell was stable for more than 5,900 hours. The proposed strategy, when experimented with using other carbon-supported metal catalysts, also gave improved results in activity and stability.
Outlook
The main shortcoming of DEFCs running in the alkaline condition is their durability. Currently, it is not sufficient for practical applications. Moreover, the anion-exchange membranes in use have two issues:
- Structural stability of membrane is insufficient for long-term use
- Carbonation occurs in presence of CO2 due to its reaction with hydroxide ions, ultimately degrading the catalyst.
Albeit stable for remarkable 5,900 hours, the membrane was replaced after 1,200 hours in the presented study. Since replacing membranes require complete disassembly of the cell, this is not a long-term practical solution.
Hence, there must be further research on increasing ionic conductivity and stability of anionic membranes for practical use of DEFC in alkaline conditions. Ideally, the hydroxide solution used to increase ionic conductivity is avoided to preserve energy density and reduce the complexity of the device. Solid oxide fuel cells offer a solution for these problems since the fuel is oxidized in gaseous form but their ceramic membrane are too fragile for mobile applications.
The current experiment makes significant strides in improving power density in DEFCs much more than any state-of-the-art DEFCs. The way ahead is further research to overcome these smaller obstacles in the long-term use of anionic membranes.
Experimental analysis
Materials used
Commercial Pd/C (10%, 8 nm Pd particles on activated carbon), as well as Pt/C (20%, 3 nm Pt particles on carbon black), were used as baseline catalysts. Also, Nafion™ solution (5%), carbon paper (TGP-H-060), and anion-exchange membranes (Fumasep FAS-PET-75)
Synthesis of heteroatom X-doped carbon (X–C, X=N, P, S, B, F)
Carbon black with abundant oxygen functional groups and melamine (C3H6N6) were mixed and ground, and finally pyrolyzed. After cooling to room temperature, N–C was obtained by washing with ethanol and water. The same method was used to synthesize P–C, S–C, B–C, and F–C from sodium hypophosphite anhydrous, sulfur powder, boric acid, and polyvinylidene difluoride.
Synthesis of hetero-atom fluorine-doped carbon catalysts
N–C and polyvinylidene difluoride were mixed and ground before adding them into a solution of acetone and water. After ultra sound treatment, the mixture was refluxed in an oil bath until fully dried. The mixture was then pyrolyzed and after cooling to room temperature, the samples were washed with ethanol and ultrapure water, followed by a vacuum to obtain the fluorinated catalyst support. The same method was used for the other precursors.
A microwave reduction method was used to synthesize palladium catalyst on the catalyst support. The content of palladium in all samples was kept at 1.0%, which was determined and double-confirmed by X-ray spectroscopy and inductively coupled plasma.
Electrochemical characterizations
For the electrical measurements, either a glassy carbon ring-disc electrode or rotating ring-disc electrode were used. The Fumasep membrane was used as an anion-exchange membrane, modified to change it to a hydroxide environment.
Reference
Chang et al., 2021, Improving Pd–N–C fuel cell electrocatalysts through fluorination-driven rearrangements of local coordination environment. Nature Energy 6, 1144–1153 https://doi.org/10.1038/s41560-021-00940-4
Image Source: P_Wei, Pixabay