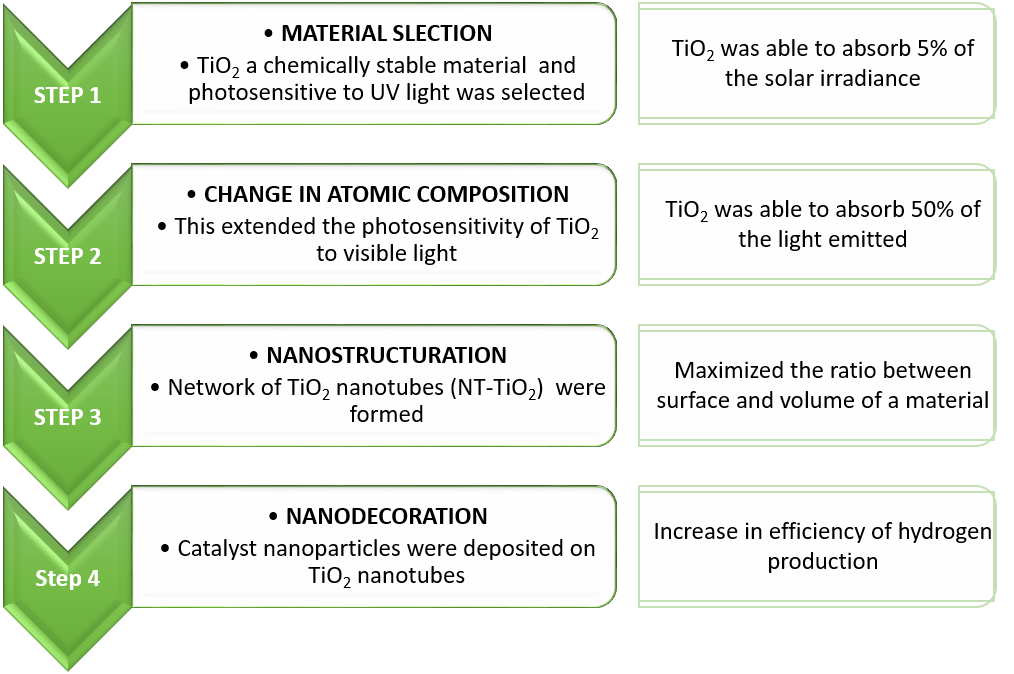
Polymer electrolyte membrane (PEM) fuel cells have high power density, low operational temperatures. If PEM runs on green hydrogen, it doesn’t even emit carbon. But their fabrication requires perfluorinated sulfonic acid (PFSA) polymers as an electrolyte separator membrane and as an ionomer in the electrode, which is quite expensive. Nafion® is the leading commercial PFSA polymer in the market. However, its manufacturing is costly as well as environmentally hazardous. Therefore, low-cost, environmentally friendly PFSA polymer substitutes are the primary goals for the fuel cell scientific community worldwide.
Researchers of the Texas A&M University and Kraton Performance Polymers Inc. experimented using NEXAR ™ polymer membranes in hydrogen fuel cells, studying different ion exchange capacities. NEXAR ™ polymer membranes are commercially available sulfonated pentablock terpolymers. They published the results in the Journal of Membrane Science . Previous studies showed that changing the ion exchange capacity, that is, the degree of sulfonation of NEXAR ™ membranes can alter the nanoscale morphology and significantly affect mechanical properties. This may influence fuel cell performance. Hence, this polymer may be used as a membrane alternative to Nafion® in fuel cells.
Experimental procedure
- Materials under consideration: three different variants of the polymer were taken up each with different Ion Exchange Capacities (IECs: 2.0, 1.5, and 1.0 meq / g), which were named NEXAR ™ -2.0, NEXAR ™ -1.5, and NEXAR ™ – 1.0 respectively.
- NEXAR ™ membrane preparation: NEXAR ™ membranes were fabricated by casting the NEXAR ™ solutions onto a silicon-coated Mylar PET film using an automatic film applicator under ambient conditions. Two different sizes were manufactured for measuring mechanical properties and conductivity.
- NEXAR ™ membrane characterization: mechanical properties using the size 25 mm (L) x 0.5 mm (W) membrane as test pieces were determined and for proton conductivity test pieces of size 30 mm (L) x 10 mm (W) were tested upon.
- Nafion® electrode fabrication: conventional Nafion® electrodes were also fabricated as controls to conduct simultaneous tests.
- NEXAR ™ electrode fabrication: NEXAR ™ electrodes were prepared in 2 ways for the 2-part study, each with a different composition.
- Electrode characterization: electrode profiling was done using a scanning electron microscopy (SEM).
- Membrane electrode assembly (MEA) and fuel cell tests: MEAs were fabricated by placing the membrane in between two catalyst-coated gas diffusion layers (anode and cathode) and heat pressing. The entire fuel cell assembly consisted of an MEA, two gaskets, and two flow plates placed between copper current collectors followed by end plates all held together by bolts. Fuel cell performance tests were conducted under ambient pressure with saturated (100% RH) anode and cathode flow rates of 0.43 L / min hydrogen and 1.02 L / min oxygen respectively.
- Electrochemical impedance spectroscopy (EIS): electrochemical impedance spectroscopy was performed after the fuel cell tests and the results analyzed.
Results
NEXAR ™ -2.0 and NEXAR ™ -1.5 had a similar proton conductivity at all temperatures, suggesting that there is a maximum limit in proton conductivity. On the contrary, NEXAR ™ membranes, when compared to Nafion® NR-212 membranes , have sufficient proton conductivity to translate into high power density hydrogen fuel cell performance.
However, NEXAR ™ -2.0 and NEXAR ™ -1.5 membranes (with Nafion® as Ionomer) did not exhibit expected fuel cell performance at all fuel cell operating conditions (temperature, pressure, voltage and humidity). Surprisingly, the NEXAR ™ -1.0 membrane (with Nafion® as Ionomer) showed expected fuel cell performance across all fuel cell operating conditions and comparable power densities to Nafion® , suggesting that NEXAR ™ -1.0 may be a viable alternative to Nafion® in hydrogen fuel cells.
During fuel cell operation the NEXAR ™ -1.0 / NEXAR ™ -1.0 membrane-ionomer was thermally and mechanically stable. These results were supported by the power density results, where MEAs with NEXAR ™ -1.0 membrane-ionomers performed better than all the other MEAs.
From the above-mentioned results it became evident that the NEXAR ™ -1.0 variant was the optimal contender to substitute current state-of-the-art PFSA polymers.
Further, to understand the impact of the NEXAR ™ -1.0 ionomer on fuel cell performance, the composition of the ionomer and solvent mixture ratios in the catalyst ink solution were modified and investigated. Results suggested that NEXAR ™ -1.0 as an ionomer behaves similarly to Nafion® ionomers in fuel cell electrodes.
SEM analysis suggested that the amount of ionomer has a significant impact on the binding of ionomer to the catalyst particles, and consequently on the catalyst layer morphology. Therefore, there is an optimum catalyst / ionomer ratio of 2/1 for the Pt / C ionomer using NEXAR ™ -1.0 in fuel cell electrodes.
Conclusions
Ultimately, NEXAR ™ -1.0 is a potentially commercially viable greener substitute to Nafion® as a membrane and ionomer in PEM Fuel cell applications due to its high conductivity, however; alternative block compositions may improve the properties of the polymer to minimize resistances within the fuel cell to match the performance of Nafion® .
Overall Nafion® / Nafion® MEAs still showed the highest fuel cell performance when overall performance was taken into account but alternative hydrocarbon-based polymer compositions for the NEXAR ™ Polymer might provide a future non-fluorinated polymer as a Nafion® substitute for PEM fuel cells .
Way forward
More analysis is required to perhaps get an accurate approximation of what variant of the NEXAR ™ polymer might cut the mark, future research may be focused upon exploring variants of Ion Exchange Capacities ranging from say 1 meq / g to 1.5 meq / g. But for now, it can be said that NEXAR ™ polymer shows promise as a viable replacement as a non-fluorinated membrane, and perhaps further research with more iterations of mechanical specs as well as chemical specs of the material we might witness a breakthrough.
Reference: https://doi.org/10.1016/j.memsci.2021.119330 : Sulfonated pentablock terpolymers as membranes and ionomers in hydrogen fuel cells , Journal of Membrane Science, 2021, 119330